
Perjeta(pertuzumab)注射剂使用说明书2012年6月第一版
批准日期:2012年6月8日;公司:Genentech
http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125409lbl.pdf
美国食品药品监督管理局(FDA)的药物评价和研究中心主任Janet Woodcock,M.D.说:“对转移乳癌需要另外治疗,今天我们做出决定批准此药而不延迟患者的可供利用性与未来供应相关的未解决生产问题”“Genentech当前正在发展一个计划以减轻患者短缺Perjeta的任何潜在影响。”
FDA的药物评价和研究中心血液学和肿瘤产品办公室主任RichardPazdur, M.D.说“自从曲妥单抗第一个被批准十年以上前,继续研究已允许我们更好了解HER2在乳癌中的作用,”“这项研究提供结合两个靶向药物结合的背景 – 曲妥单抗和Perjeta与多西他奇[docetaxel]减慢乳癌疾病进展。”
Perjeta正在被批准有一个黑框警告警告患者和卫生保健专业人员对胎儿死亡或严重效应的潜在风险。开始Perjeta治疗前必须证实妊娠状态。
处方资料重点
这些重点不包括安全和有效使用PERJETA所需所有资料。请参阅下文为PERJETA的完整处方资料
PERJETATM (pertuzumab)注射剂,为静脉使用
美国初次批准:2012

1适应证和用途
PERJETA是一种HER2/neu受体拮抗剂适用于与曲妥单抗[trastuzumab]和多西他奇[docetaxel]联用为未曾接受既往抗-HER2治疗或化疗的HER2-阳性转移乳癌患者为转移疾病的治疗。
2剂量和给药方法
(1)只为静脉输注。不要静脉推注或丸注给药。(2.3)
(2)初始剂量为840 mg历时60-分钟静脉输注。其后每3周420 mg历时30至60分钟静脉输注。(2.1)
3剂型和规格
420 mg/14 mL单次用小瓶。(3)
4禁忌证
无。(4)
5警告和注意事项
(1)胚胎-胎儿毒性:但给予妊娠妇女可能发生胎儿危害。(5.1, 8.1)
(2)左心室功能不全:监视LVEF和如适当时撤消给药。(5.2, 6.1)
(3)输注相关反应,超敏性反应/过敏反应:监视体征和症状。如发生重要输注-相关反应,减慢或中断输注和给予适当医药治疗。(5.3)
(4)HER2测试:由证实精通熟练实验室用FDA批准的检验进行。(5.4)
6不良反应
用PERJETA与曲妥单抗和多西他奇联用最常见不良反应(>30%)是腹泻,脱发,中性细胞减少,恶心,疲乏,皮疹,和周围神经病。(6.1)
报告怀疑不良反应,联系Genentech电话1-888-835-2555或FDA电话1-800-FDA-1088或www.fda.gov/medwatch。
7在特殊人群中使用
(1)哺乳母亲:终止哺乳或终止PERJETA,考虑药物对母亲的重要性。(8.3)
(2)女性的生殖潜能:忠告女性关于预防妊娠和计划,鼓励患者参加MotHER妊娠注册电话1-800-690-6720联系。(5.1,8.1, 8.6, 17)
完整处方资料

1适应证和用途
PERJETA适用于与曲妥单抗和多西他奇联用为治疗未曾接受既往抗-HER2治疗或化疗为转移疾病HER2-阳性转移乳癌患者。
2 剂量和给药方法
2.1 推荐剂量和方案
PERJETA的初始剂量是840 mg历时60-分钟静脉输注给药,其后每3周剂量420 mg历时30至60分静脉输注。
当用PERJETA给药时,推荐的曲妥单抗初始剂量为8 mg/kg历时90-分钟静脉输注,其后每3周剂量6mg/kg历时30至90分静脉输注。
当用PERJETA给药时,推荐的多西他奇初始剂量是75 mg/m2静脉输注,如初始剂量耐受良好时剂量可扩增至每3周100 mg/m2。
2.2剂量调整
对延迟或丢失剂量,如两次顺序输注间的时间小于6周,应给予420mg剂量PERJETA。不要等待直至下一次计划的剂量。如两次顺序输注间的时间是6周或以上,应再次给予840 mgPERJETA初始剂量历时60-分钟静脉输注,其后每3周剂量420 mg历时30至60分静脉输注给药。
如患者发生输注-相关反应,PERJETA的输注速率可减慢或中断。如患者经受严重超敏反应应立即终止输注[见警告和注意事项(5.2)]。
左心室射血分数(LVEF):
对以下任何一种情况不给PERJETA和曲妥单抗给药至少3周:
(1)LVEF下降至小于40%或
(2)LVEF为40%至45%与绝对下降低至治疗前值10%或更大[见警告和注意事项(5.2)]
如LVEF已恢复至大于45%或至40%至45%伴随绝对下降小于低至治疗前值10% PERJETA可恢复给药。
如约3周内重复评估后,LVEF没有改善,或进一步下降,应强烈考虑终止PERJETA和曲妥单抗,除非对个体患者的获益被认为胜过风险[见警告和注意事项(5.2)]。
如曲妥单抗治疗不给予或终止时,PERJETA应不给予或终止。
如果多西他奇被终止,可继续用PERJETA和曲妥单抗治疗。
不建议减低PERJETA剂量。
对多西他奇调整剂量,请见多西他奇处方资料。
2.3给药准备
只为静脉输注给药。不要静脉推注或丸注给药。
PERJETA不要与其它药物混合。
制备
(1)用无菌术制备输注溶液如下:
(2)非肠道药物给药前应肉眼观测有无颗粒物和变色。
(3)从小瓶抽吸适当容积的溶液。
(4)稀释至250 mL 0.9% 氯化钠PVC或非-PVC聚烯烃输液袋。
(5)轻轻倒置混合稀释液。不要摇动。
(6)一旦制备好立即给药。
(7)如果稀释的输注溶液不立即使用,它可贮存在2°C至8°C至24小时。
(8)只用0.9%氯化钠注射液稀释。不要使用葡萄糖(5%)溶液。
3剂型和规格
PERJETA (pertuzumab) 420 mg/14 mL (30 mg/mL)在单次用小瓶中。
4禁忌证
无。
5警告和注意事项
5.1胚胎-胎儿毒性
当给予妊娠妇女PERJETA可致胎儿危害。妊娠食蟹猴用治疗pertuzumab导致羊水过少,延迟胎儿肾发育,和胚胎-胎儿死亡。如妊娠期间给予PERJETA,或如当接受本药患者成为妊娠,应忠告患者对胎儿的潜在危害[见在特殊人群中使用(8.1)]。
PERJETA开始前确认妊娠状态。忠告患者胚胎-胎儿死亡和出生缺陷风险和治疗时和后需要避孕。忠告患者如她们怀疑可能妊娠时立即联系她们的卫生保健提供者。如果妊娠期间或如患者接受PERJETA时成为妊娠如果给予PERJETA,立即报告暴露至Genentech不良事件线电话1-888-835-2555。鼓励妊娠期间被暴露妇女通过联系电话1-800-690-6720纳入MotHER妊娠注册,对孕龄和符合社区医护标准适当的进行胎儿测试。羊水过少处理中静脉水化的疗效由于PERJETA暴露不知道。
5.2左心室功能不全
在随机试验中曾报道用阻断HER2活性药物,包括PERJETA减低LVEF。PERJETA与曲妥单抗和多西他奇联用与安慰剂与曲妥单抗和多西他奇联用比较,不伴随症状性左心室收缩功能障碍(LVSD)发生率增加或LVEF降低[见临床研究(14.1)]。PERJETA-治疗组患者中左心室功能不全发生4.4%和安慰剂-治疗组患者8.3%。PERJETA-治疗组患者中症状性左心室收缩功能障碍(充血性心脏衰竭)发生1.0%和安慰剂-治疗组患者1.8%[见不良反应(6.1)]。曾接受既往蒽环类药物[anthracyclines]或既往对胸区放疗患者可能处在降低LVEF较高的风险。
未曾在治疗前LVEF值≤ 50%患者中,既往CHF史,既往曲妥单抗治疗期间LVEF减低至<50%,或可能损伤左心室功能情况例如未控制高血压,近期心肌梗死,严重心率失常需要治疗或既往蒽环类药物累计暴露至>360 mg/m2阿霉素[doxorubicin]或其等同物中研究PERJETA。
PERJETA开始前和治疗期间定期(如,每三个月)评估LVEF以确保LVEF在机构正常限度。如果LVEF是<40%,或是40%至45%与或绝对降低较大低于治疗前值10%时,不用PERJETA和曲妥单抗和约3周内重复LVEF评估。如果LVEF无改善或进一步下降终止PERJETA和曲妥单抗,除非对个体患者获益胜过风险[见剂量和给药方法(2.2)]。
5.3输注相关反应,超敏性反应/过敏反应
PERJETA曾伴随输注和超敏性反应[见不良反应(6.1)]。输注反应被定义在随机试验中任何事件被描述为超敏性,过敏反应,急性输注反应或细胞因子释放综合征发生在输注时或输注相同天。PERJETA的初始剂量是给予在曲妥单抗和多西他奇前1天允许检查伴PERJETA-反应。在第一天,当只给予PERJETA,PERJETA-治疗组输注反应的总体频数为13.0%和安慰剂-治疗组9.8%,小于1%是3或4级。最常见输注反应(≥1.0%)是发热,寒战,疲乏,头痛,虚弱,超敏性,和呕吐。
在第二个疗程时当所有药物都在相同天给药,PERJETA-治疗组最常见输注反应(≥1.0%)是疲乏,味觉障碍,超敏性,肌肉痛,和呕吐。
在随机试验中PERJETA-治疗组的超敏性/过敏反应总体频数是10.8%和安慰剂-治疗组9.1%。PERJETA-治疗组3 –4级超敏性/过敏反应发生率为2%和安慰剂-治疗组2.5%按照美国国家癌症研究所–对不良事件常见名词标准(NCI - CTCAE)(版本3)。总之,4/124例PERJETA-治疗组患者和安慰剂-治疗组 2例患者经受过敏反应。
第一次输注后密切观察患者60分钟和随后PERJETA输注后30分。如发生重要输注-相关反应,输注缓慢或中断和给予适当医药治疗。小心仔细监视患者直至体征和症状完全解决。有严重输注反应患者考虑永远终止[见剂量和给药方法(2.2)]。
5.4HER2测试
为选择患者适当为PERJETA治疗需要检测HER2蛋白过表达因为这些是唯一的研究患者和曾显示获益[见适应证和用途(1)和临床研究(14)]。在随机试验中,乳癌患者需要有HER2过表达的证据,被定义用DakoHerceptest™为3+IHC或用Dako HER2 FISH PharmDx™检验药盒FISH扩增比值≥2.0。对患者其乳癌用FISH是阳性,但用IHC不显示蛋白过表达只能得到有限数据。
HER2状态的评估应由证实正在用特殊技术精通熟练实验室进行。不适当分析性能,包括使用低于最优固定组织,不使用指定试剂,偏离特殊分析指导,和不能包括确证分析的适当对照,可能导致不可靠结果。
6不良反应
在说明书其它节内更详细讨论下列不良反应:
(1)胚胎-胎儿毒性[见警告和注意事项(5.1)]
(2)左心室功能不全[见警告和注意事项(5.2)]
(3)输注相关反应,超敏性反应/过敏反应[见警告和注意事项(5.3)]
6.1临床试验经验
因为临床试验是在广泛不同情况下进行的,临床试验观察到不良反应率不能与另一种药临床试验发生率直接比较而且可能不反映实践中观察到的发生率。
在临床试验中,曾在1400例以上有各种恶性病和用PERJETA治疗主要与其它抗肿瘤药物联用患者中评价PERJETA。
在表1描述的是在随机试验中在804例用HER2-阳性转移乳癌治疗患者确定的不良反应。患者随机接受或PERJETA与曲妥单抗和多西他奇联合或安慰剂与曲妥单抗和多西他奇联合。PERJETA-治疗组患者研究治疗中位时间为18.1个月和安慰剂-治疗组患者11.8个月。对PERJETA或曲妥单抗不允许调整剂量。PERJETA-治疗组患者不良事件率导致永远终止所有研究治疗为6.1%和安慰剂-治疗组为5.3%患者。PERJETA-治疗组因不良事件导致单独终止多西他奇23.6%患者和安慰剂-治疗组为23.2%患者。表1报道了PERJETA-治疗组至少10%患者发生的不良反应。
PERJETA与曲妥单抗和多西他奇联用最常见不良反应(>30%)是腹泻,脱发,中性细胞减少,恶心,疲乏,皮疹,和周围神经病。最常见NCI-CTCAE(版本3)3 – 4级不良反应(>2%)为中性细胞减少,发热中性细胞减少,白细胞减少,腹泻,周围神经病,贫血,虚弱,和疲乏。两治疗组中均观察到亚裔和来自其它地理区域患者比其它种族患者发热中性细胞减少发生率增加。亚裔患者中,发热中性细胞减少发生率pertuzumab-治疗组(26%)与安慰剂-治疗组(12%)比较较高。
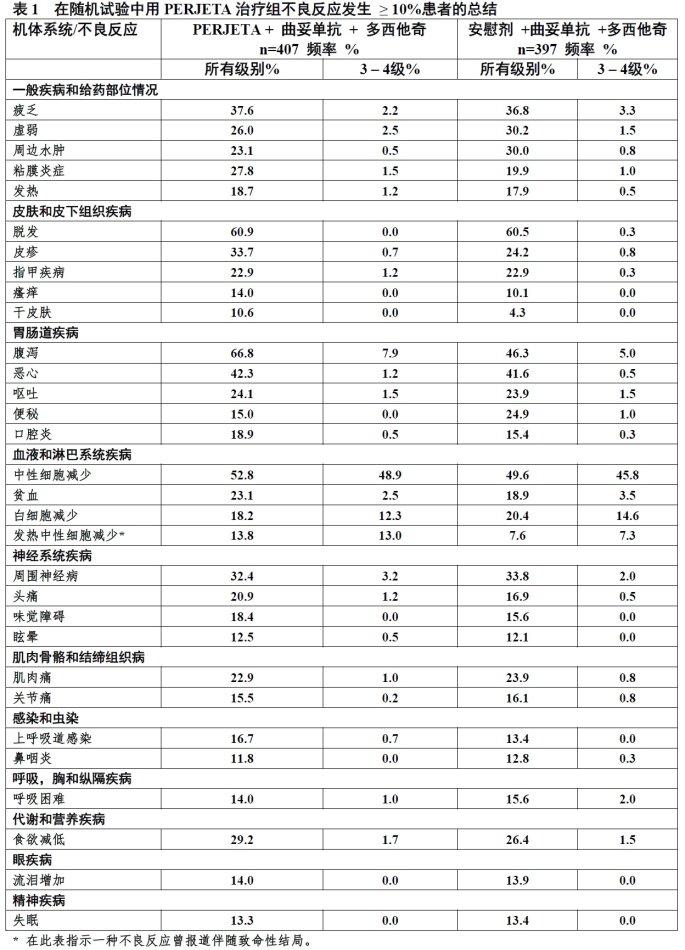
在PERJETA-治疗组中<10%患者报道以下临床相关不良反应:
皮肤和皮下组织疾病:甲沟炎(PERJETA-治疗组7.1%相比安慰剂-治疗组3.5%)
呼吸,胸和纵隔疾病:熊膜积液(PERJETA治疗组5.2%相比安慰剂-治疗组5.8%)
心脏病:左心室功能不全(PERJETA-治疗组4.4%相比安慰剂-治疗组8.3%)包括症状性左心室功能障碍(CHF)(PERJETA-治疗组1.0%相比安慰剂-治疗组1.8%)
免疫系统疾病:超敏性(PERJETA-治疗组10.1%相比安慰剂-治疗组8.6%)
终止多西他奇后接受PERJETA和曲妥单抗患者中报道的不良反应
在随机试验中,终止多西他奇治疗后报道的不良反应频数较少。在PERJETA和曲妥单抗治疗组中所有不良反应发生<10%患者有除了腹泻(19.1%),上呼吸道感染(12.8%),皮疹(11.7%),头痛(11.4%),和疲乏(11.1%)。
6.2免疫原性
如同所有治疗性蛋白,有对PERJETA免疫反应的潜能。
在随机试验中患者在多个时间点检测对PERJETA抗体。PERJETA-治疗组约2.8%(11/386)患者和安慰剂-治疗组6.2%(23/372)患者检出阳性抗-PERJETA抗体。这些34例患者中,没有1例经受过敏反应/超敏性反应是明确与抗-治疗性抗体(ATA)相关。患者血清中pertuzumab的存在其水平预期在ATA采样的时间可能干扰分析检出抗pertuzumab抗体的能力。此外,分析可检测对曲妥单抗的抗体。作为结果,数据可能不能准确地反映抗-pertuzumab抗体发生真实的发生率。
免疫原性数据是高度依赖于所有测试方法灵敏度和特异性,此外,Additionally,在一种测试方法观察到的阳性结果发生率可能受几种因子影响,包括样品处理,采样时间,药物干扰,同时用药,和所患疾病。因为这些理由,比较对PERJETA抗体的发生率与对其它产品抗体发生率可能是误导。
7药物相互作用
未观察到pertuzumab和曲妥单抗间,或pertuzumab和多西他奇间药物-药物相互作用。
8在特殊人群中使用
8.1妊娠
妊娠类别D
风险总结
在妊娠妇女中没有PERJETA的适当和对照良好研究。根据动物研究发现,当给予PERJETA至妊娠妇女可致胎儿危害。妊娠所有3个三个月期间可能存在PERJETA的影响。根据Cmax在推荐人剂量临床上相关暴露大于2.5至20-倍时,妊娠食蟹猴给予Pertuzumab导致羊水过少,延迟胎儿肾发育,和胚胎-胎儿死亡。如果妊娠期间给予PERJETA,或如患者当接受PERJETA时成为妊娠,应忠告患者对胎儿潜在危害。
如妊娠期间给予PERJETA或如患者当接受PERJETA时成为妊娠,立即报告暴露至Genentech不良反应线电话1-888-835-2555。鼓励妊娠期间可能被暴露妇女纳入MotHER妊娠注册通过联系电话1-800-690-6720[见患者咨询资料(17)]。
动物数据
在食蟹猴中曾进行生殖毒理学研究。妊娠猴在怀孕天(GD)19用负荷剂量30至150 mg/kgpertuzumab处理,接着每两周剂量10至100 mg/kg。这些剂量水平导致临床上相关暴露,根据Cmax大于推荐人用剂量2.5至20-倍,静脉给予pertuzumab从怀孕GD19至GD50(器官形成期)是胚胎毒性,GD25至GD70间有依赖剂量的胚胎-胎儿死亡增加。每两周用pertuzumab剂量10,30,和100 mg/kg(根据 Cmax大于推荐人用剂量2.5至20-倍)处理的母兽胚胎-胎儿丢失发生率,分别为33,50,和85%。在怀孕GD100剖腹产,羊水过少,肺和肾重相对减低和在所有pertuzumab剂量组确定肾发育不全的显微镜证据符合肾发育延迟。所有处理组的子代报道Pertuzumab暴露,母体血清水平在怀孕GD100时的29%至40%。
8.3哺乳母亲
不知道PERJETA是否排泄至人乳汁中,但人IgG被排泄在人乳汁中。因为许多药物是排泄在人乳汁和因为在哺乳婴儿来自PERJETA严重不良反应的潜能,应做出决策是否终止哺乳,或终止药物,考虑到PERJETA的半衰期和药物对母亲的重要性[见警告和注意事项(5.1),临床药理学(12.3)]。
8.4儿童使用
尚未确定在儿童患者中PERJETA的安全性和有效性。
8.5老年人使用
在随机试验中接受PERJETA的402例患者中,60例患者(15%)是 ≥ 65岁和5例患者(1%)是 ≥75岁。未观察到这些患者和较年轻患者间PERJETA的疗效和安全性总体差别。
根据一项群体药代动力学分析,未观察到pertuzumab药代动力学显著差异。
8.6女性的生殖潜能
当妊娠期间给予PERJETA可致胚胎-胎儿危害。忠告患者关于防止和计划妊娠。建议有生殖潜能女性当接受PERJETA和末次剂量PERJETA后6个月时使用有效避孕。
如妊娠期间给予PERJETA或如患者接受PERJETA时成为妊娠,立即报告暴露至Genentech不良事件线电话1-888-835-2555。鼓励妊娠期间可能被暴露妇女纳入MotHER妊娠注册通过联系电话1-800-690-6720[见患者咨询资料(17)]。
8.7肾受损
有轻度(肌酐清除率[CLcr]60至90 mL/min)或中等(CLcr 30至60mL/min)肾受损患者无需调整PERJETA剂量。因为对严重肾受损患者(CLcr小于30mL/min)可供利用有限的药代动力学数据可能无剂量调整的建议[见临床药理学(12.3)]。
8.8肝受损
未曾进行临床研究评价肝受损对pertuzumab药代动力学的影响。
10药物过量
迄今无用PERJETA 药物过量报道。
11一般描述
Pertuzumab是一种重组人源化单抗,靶向细胞外人表皮生长因子受体2蛋白(HER2)二聚化结构区(子结构区II)。Pertuzumab通过重组DNA技术在一种哺乳动物细胞(中国仓鼠卵巢)含抗生素,庆大霉素培养中生产。最终产品中不能检测到庆大霉素。Pertuzumab分子量约148kDa。
PERJETA是一种无菌,透明至轻度乳白色,无色至浅棕色液体为静脉输注。每个单次使用小瓶含420 mg的pertuzumab在浓度30mg/mL在20 mM L-组氨酸醋酸盐(pH 6.0), 120 mM蔗糖和0.02%聚山梨醇20。
have been conducted
12临床药理学
12.1 作用机制
Pertuzumab靶向细胞外人表皮生长因子受体2蛋白(HER2)的二聚化结构区(子结构区II),因此,阻断HER2与其它HER家族成员,包括EGFR,HER3和HER4,配基-依赖性异源二聚化作用。其结果,pertuzumab抑制配体-启动细胞内信号通过两条主要信号通路,丝裂原-激活的蛋白(MAP)激酶和磷酸肌醇3-激酶(PI3K)。这些信号通路的抑制分别导致细胞生长停止和凋亡。此外,pertuzumab介导抗体-依赖细胞-介导细胞毒性(ADCC)。
而单独pertuzumab抑制人肿瘤细胞增殖,在HER2-过表达外移植肿瘤模型中pertuzumab和曲妥单抗的联用显着增强抗肿瘤活性。
12.3 药代动力学
剂量范围2 – 25 mgkg时Pertuzumab显示线性药代动力学。根据一项群体PK分析包括481例患者,pertuzumab的中位清除率(CL)为0.24L/day和中位半衰期是18天。用初始剂量840 mg随后维持剂量420 mg每三周其后,在首次维持剂量后达到pertuzumab稳态浓度。
群体PK分析提示根据年龄,性别,和种族(日本人相比非-日本人)无PK差别。基线血清白蛋白水平和瘦体重为协变量对PK参数只发挥次要影响。所以,无需根据体重或基线白蛋白水平调整剂量。
在随机试验中在一项37例患者子-研究未观察到pertuzumab和曲妥单抗间,或pertuzumab和多西他奇间药物-药物相互作用。
对PERJETA未曾进行致力于肾受损试验。根据群体药代动力学分析的结果,在患者有轻度(CLcr 60至90mL/min,n=200)和中等肾受损(CLcr 30至60 mL/min,n=71)pertuzumab暴露与正常肾功能患者(CLcr大于90 mL/min, n=200)相似。
在CLcr(27至244 mL/min)范围未观察到CLcr和pertuzumab暴露间相互关系。
12.6 心脏电生理学
在随机试验中在20例HER2-阳性乳癌患者子组中评价用初始剂量pertuzumab 840 mg后每三周用维持剂量420mg对QTc间期的影响。试验中根据Fridericia校正方法在平均QT间期(肌,大于20ms)与安慰剂未检测到大变化,因为试验设计限制不能除外平均QTc间期小增加(即,小于10 ms)。
13非临床毒理学
13.1 癌发生,突变发生,生育能力受损
未曾在在动物中进行长期研究评价pertuzumab的致癌潜能。
未曾进行研究评价pertuzumab的致突变潜能。
在动物中未曾进行特殊生育能力研究评价pertuzumab的影响。在食蟹猴重复给药至6个月时间毒性研究中未观察到对雄性和雌性生殖器官的不良影响。
14临床研究
14.1 转移乳癌
随机试验是一项808例HER2-阳性转移乳癌患者的多中心,双盲,安慰剂-对照试验。要求乳腺肿瘤标本显示HER2过表达,被定义为在一个中心实验室测定3+IHC或FISH的扩增比值≥2.0。患者被随机化1:1接受安慰剂加曲妥单抗和多西他奇或PERJETA加曲妥单抗和多西他奇。按既往治疗(既往有或既往无辅助/新辅助抗-HER2治疗或化疗)和地理区域(欧洲,北美,南美,和亚洲)随机化分层。有既往辅助或新辅助治疗患者要求在纳入试验前有无疾病时间间隔大于12个月。
静脉给予PERJETA初始剂量840 mg。接着是其后每3周420 mg。静脉给予曲妥单抗初始剂量8 mg/kg,接着其后每3周6mg/kg。患者用PERJETA和曲妥单抗治疗直至疾病进展,知情,或不能接受的毒性撤药。每3周静脉输注给予多西他奇初始剂量75mg/m2至少6疗程。如初始剂量耐受良好在研究者裁决下多西他奇剂量可扩增至100mg/m2。在主要分析时,安慰剂-治疗组给予研究治疗疗程的平均数为16.2个月和PERJETA-治疗组为19.9个月。
随机试验的主要终点是由独立评审机构(IRF)评估无进展活存(PFS)。PFS被被定义为随机化日期至疾病进展或(任何原因)死亡的日期,如死亡发生在18个月最末次肿瘤评估内。另外终点包括总活存(OS),PFS(研究者-评估),客观缓解率(ORR)和缓解时间。
治疗组间患者人口统计指标和基线特征被平衡。中位年龄为54岁(范围22至89岁),59%为白人,32%为亚裔,和4%为黑人。除2例患者所有是妇女。70%患者在北美纳入,14%在南美,38%欧洲,和31%亚洲。在研究组中肿瘤预后特征,包括激素受体状态(阳性48%,阴性50%),存在内脏疾病(78%)和非-内脏疾病仅(22%)相似。约半数患者接受既往辅助或新辅助抗-HER2治疗或化疗(安慰剂47%,PERJETA46%)。有激素受体阳性肿瘤患者中,45%接受既往辅助激素治疗和11%为转移疾病接受激素治疗。11%患者接受既往辅助或新辅助曲妥单抗。
随机试验证实在PERJETA-治疗组与安慰剂-治疗组比较独立评审机构IRF-评估PFS统计显著改善[危害比 (HR) =0.62(95% CI: 0.51, 0.75), p <0.0001]和中位PFS增加6.1个月(PERJETA-治疗组中位PFS为18.5个月相比安慰剂-治疗组12.4个月)(见图1)。研究者-评估PFS结果与独立评审机构IRF-评估PFS有可比性。
跨越几个子组患者包括年龄(< 65或≥65岁),种族,地理区域,既往辅助/新辅助抗-HER2治疗或化疗(是或否),和既往辅助/新辅助曲妥单抗(是或否)观察到一致结果。激素受体-阴性病(n=408)子组患者危害比为0.55(95%CI: 0.42, 0.72)。激素受体-阳性疾病患者子组中(n=388),危害比0.72(95% CI: 0.55,0.95)。在有病患者子组中限于非内脏转移(n=178),危害比为0.96(95% CI: 0.61, 1.52)。
在PFS分析时,165例患者已死亡。安慰剂治疗组(23.6%)与PERJETA-治疗组(17.2%)比较发生更多死亡。中期OS分析时。结果不够成熟和不能符合预先指定停止对统计显著性的边界。见表2和图2。


图1对随机试验独立评审机构-评估无进展活存的Kaplan-Meier曲线
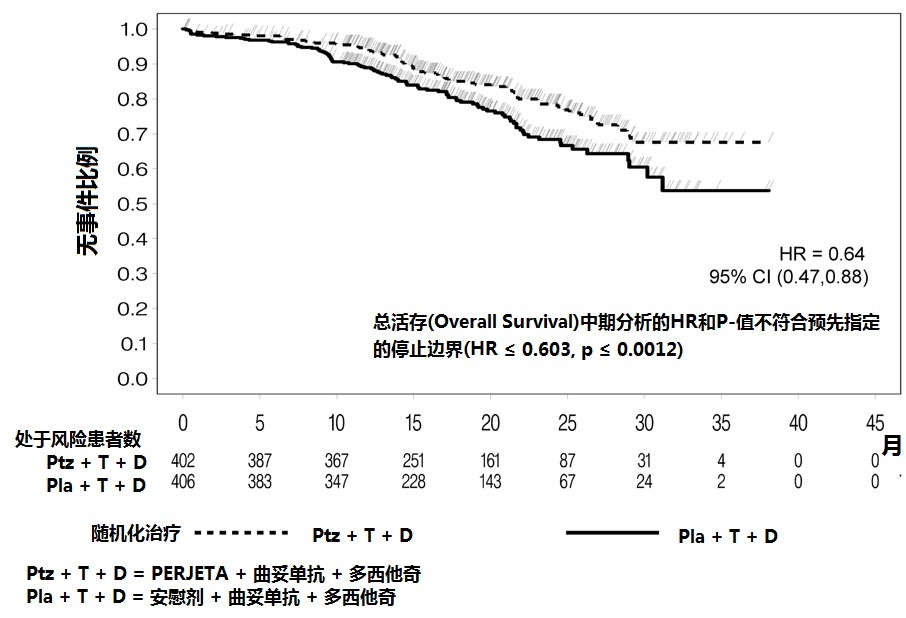
图2随机化试验总活存的Kaplan-Meier曲线
16如何供应/贮存和处置
16.1 如何供应
PERJETA以一个420 mg/14 mL(30 mg/mL)单次用小瓶无防腐剂溶液供应。NDC50242-145-01。
用前小瓶贮存在冰箱在2°C至8°C(36°F至46°F)。
为了避光保护小瓶在外面纸盒中。
17患者咨询资料
(1)忠告妊娠妇女和有生育能力女性PERJETA暴露可能导致胎儿危害,包括胚胎-胎儿死亡或出生缺陷[见警告和注意事项(5.1)和在特殊人群中使用(8.1)]。
(2)忠告有生育能力女性当接受PERJETA和末次剂量PERJETA后6个月使用有效避孕[见警告和注意事项(5.1)和在特殊人群中使用(8.6)]
(3)忠告用PERJETA治疗哺乳母亲终止哺乳或终止PERJETA,考虑药物对母亲的重要性[见在特殊人群中使用(8.3)]。
(4)鼓励妊娠期间被暴露于PERJETA妇女纳入MotHER妊娠注册通过电话1-800-690-6720联系[见警告和注意事项(5.1)和在特殊人群中使用(8.1)。